
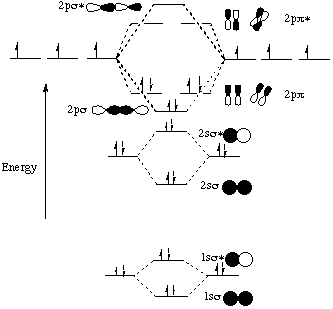
This handout is in response to questions about the energy level ordering for homonuclear diatomics. Think about the diatomic molecule N2. Each nitrogen atom has the electron configuration 1s22s22p3. These atomic electrons are shown on either side of the molecular orbital diagram. The molecular Note that the p orbitals of the two nitrogen atoms interact in two ways. Each nitrogen atom has one p orbital lying along the internuclear axis that has head-on, or s, overlap with the corresponding p orbital on the other nitrogen atom. This generates the 2ps and 2ps* molecular orbitals. Each nitrogen also has two p orbitals perpendicular to the internuclear axis that have side-on, or p, overlap with the corresponding p orbitals on the other nitrogen atom. This generates the 2pp and 2pp* molecular orbitals. They do not show as dramatic an energy difference from the nitrogen atomic orbitals because side-on overlap is not as good as head-on overlap. We can show one of the p molecular orbitals in the plane of the page; we picture the other one tilted to indicate that it is perpendicular to the plane of the page. Based on the above reasoning, we would predict that the molecular orbital energy level diagram for diatomic N2 would look something like this:
Note that all of the s bonds have cylindrical symmetry (or a C-infinity rotation element) about the internuclear axis. Note that all of the p bonds have C2 antisymmetry about the internuclear axis. Note that N2 has a bond order of 3 because there are 6 more electrons in bonding orbitals than there are in antibonding orbitals. This all makes sense so far. But actually, we know from our course reader (page III-16) that the energy level diagram for N2 is a little bit different from what we would predict. In reality, it looks like this:
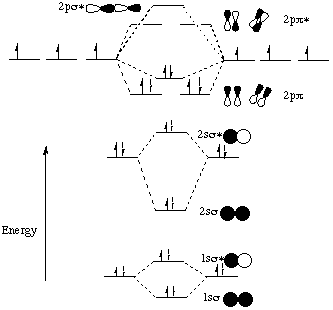
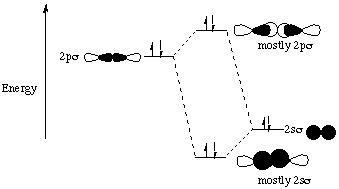
This is the same as the diagram from before except that the 2p s orbital moved up in energy while the 2ss orbital moved down in energy. (The fact that the 2ss orbital moved down in energy isn’t obvious from the energy diagram on page III-16, but trust me, it happens.) This changed the relative energies of the orbitals so that the 2ps orbital is now above the "degenerate" (same energy) 2pp orbitals. Why did this crossover happen?!The important thing to realize here is that the orbital interaction ideas we’ve been learning apply not only to atomic orbitals, but to any kind of orbital. If two orbitals 1. have the same symmetry elements in the point group of the molecule and 2. are close to each other in energy, then it is possible for them to interact. What is the point group of N2? What symmetry elements of that point group do the 2s s and 2ps molecular orbitals possess? Can these molecular orbitals interact the way atomic orbitals can? You bet they can!!When atomic orbitals interact, we show their interaction by taking + and - combinations of their relative wavefunction phases. The bonding (+) combination has the wavefunctions in phase (overlap of two shaded or two unshaded orbital lobes) and the antibonding (-) combination has the wavefunctions out of phase (a shaded orbital lobe overlapping with an unshaded orbital lobe). In the case of two molecular orbitals interacting, the same + and - combinations have to be taken. Just to be extra clear, I am going to show pictures of the two contributing molecular orbitals (2s s and 2ps) on the sides as well as pictures of their + and - ("bonding" and "antibonding") combinations in the middle of the energy diagram.
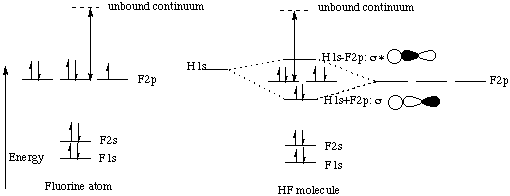
Professor Zare said in lecture that when two orbitals of different energies interact, the antibonding combination "resembles" (in terms of distribution of electron density) the higher energy interacting orbital and the bonding combination resembles the lower energy interacting orbital. Therefore, it makes sense to represent this interaction by saying that the overlap between the 2s s and 2ps molecular orbitals has the effect of pushing the 2ss orbital down in energy and pushing the 2ps orbital up in energy. Energy has to be conserved, so the amount that the 2ps orbital is pushed up has to equal the amount that the 2ss orbital is pushed down.This explains why the energy diagram for N2 has a crossover between the 2p s and 2pp molecular orbitals. The interaction between the 2ps and 2ss MO’s pushes the energy of the 2ps up above the energy of the doubly degenerate 2pp. (The 2ss is concurrently pushed down, but since the 1ss* starts out so very far below 2ss, there is no crossing of these two levels.)We are told that this crossover between the 2p s and 2pp MO’s happens for the second-row diatomics from Li2 to N2. The course reader says that for O2, F2, and Ne2, the energy diagram is as we would have predicted without taking into account the interaction between the 2ps and 2ss. Why is this the case?As we travel from left to right across a row of the periodic table, the atoms get smaller and smaller because the increased nuclear charge of the right-most elements pulls the electrons more strongly toward the center of the atom. When electrons are closer to the positively-charged nucleus, they are stabilized by their coulombic (electrostatic) attraction to the protons in the nucleus. More stable equals lower energy, so all of the orbitals decrease in energy. The orbitals that start out closer to the nucleus (i.e. lower in energy) have a larger energy decrease than the orbitals that start out further away from the nucleus (higher in energy). This is because the smaller orbital-nucleus distance results in a decreased denominator in the expression for the stabilizing Coulomb attraction between the electron and the nucleus: attraction is proportional to z1z2/r2, where the z1 is the charge of the nucleus, z2 is the charge of the electron, and r is the distance between the nucleus and the orbital containing the electron. Therefore, for our diatomic example, both the 2s s and 2ps orbitals are pulled down in energy because of the increased nuclear charge, but the 2ss orbital has a larger energy decrease than the 2ps orbital because the contributing 2s atomic orbitals penetrate closer to the nucleus than the 2p orbitals (remember, p orbitals have a node at the nucleus while s orbitals have electron density there). Thus the energy difference between the 2ss and 2ps orbitals increases. As we have seen above, if the two orbitals start too far apart in energy, it’s harder for them to interact. That’s why I made such a big deal of it on the first page of this handout: If two orbitals 1. have the same symmetry elements in the point group of the molecule and 2. are close to each other in energy, then it is possible for them to interact. If the two orbitals are not close enough in energy, then the interaction is weak or nonexistant. The idea here is that for homonuclear diatomics up to N2, the 2ss and 2ps orbitals are close enough in energy to interact strongly, but for homonuclear diatomics to the right of N2 on the periodic table, the increased nuclear charge causes the 2ss and 2ps orbitals to be energetically further apart so that the interaction is weaker. That’s why you get a crossover to the left of N2 but not to its right.Incidentally, while we’re on the subject of diatomics to the right of N2, let’s think about Ne2 for a minute. Is there a crossover in its energy diagram? How many of the molecular orbitals are filled with electrons? What is it’s bond order? Would you expect to find a diatomic molecule of neon? The ideas that we’ve gone over in this handout are going to come back to us again and again throughout chem 32. For example, look at page III-19 in your course reader. Notice that the H 1s orbital has the same symmetry elements as the F 1s and 2s orbitals within the Cƒv point group. Why then does it not interact (to any appreciable extent) with these two orbitals? The answer is that it doesn’t fulfill our second condition for interaction: the two interacting orbitals must be close in energy. The only fluorine orbital with which the H 1s orbital can interact is the F 2p orbital that points along the internuclear axis. Both of these orbitals have a Cƒ element along the internuclear axis and an infinite number of mirror planes containing the internuclear axis (those are the symmetry elements of the Cƒv point group). Since both conditions of interaction (same symmetry elements within the point group and similar energy) are fulfilled, an interaction occurs as pictured on page III-19. What happens to the two fluorine p orbitals that are perpendicular to the internuclear axis? They do not have a Cƒ element along the internuclear axis. Even without knowing this you can see that their interaction with the H 1s orbital would cancel out because there is just as much in-phase (shaded-shaded) overlap as there is out-of-phase (shaded-unshaded) overlap:
 Since there is nothing for the fluorine 2p orbitals to interact with, they just stay at the same energy as they are in the fluorine atom. This is why the first ionization energy of fluorine is the same as the first ionization energy of HF. Remember, the first ionization energy is the energy needed to pluck the highest energy electron off the atom into the unbound continuum. In the diagram below, the first ionization energy is represented by the double-headed arrow. |
|
Hopefully you are now glimpsing the beginnings of how fascinating bonding can be! Just think how exciting it’s all going to get when we start using atoms that have d orbitals too . . .
|