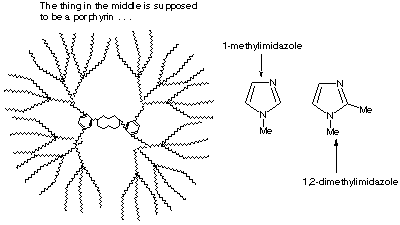
Bioinorganic 1.* (1997 F 8) Consider the dendrimer substitudted prophyrin whose structure is shown below. In separate experiments, Fe(II) and Co(II) ions were substituted into the prophyrin ligand of the dendrimer forming, in each case, neutral complexes, free of axial ligand. For the dendrimeric Fe(II) prophyrin, an excess of 1,2-dimethylimidazole was added as an axial ligand and then the equilibrium binding of O2 was determined. The P1/2(O2) value which was measured for reversible O2 binding did not change when additional 1,2-dimethylimidazole was added. In a similar experiment, an excess of 1-methylimidazole was added to the Fe(II) dendrimer porphyrin. In this case, the resulting Fe(II) porphyrin failed to bind O2 at all! Similar experiments were subsequently carried out with the Co(II) dendrimer porphyrin but the results were somewhat different. With either 1,2-dimethylimidazole or 1-methylimidazole, the Co(II) dendrimer porphyrins reversibly bound O2; two different P1/2(O2) values were obtained using 1,2-dimethylimidazole as compared with 1-methylimidazole. Explain the different behavior of the Fe(II) and Co(II) dendritic prophyrins.
A. Specifically, why did the Fe(II) porphyrin bind O2 when excess 1,2-dimethylimidazole was present but not at all when 1-methylimidazole was present? Use equations in your explanation. The sterics of the bulky diMeIm cause it to be farther below the porphyrin plane. This pulls the Fe down into the plane, making it unable to bind a second diMeIm on top (it can, however, bind the smaller O2). MeIm is not so sterically bulky and does not pull the Fe down far. Therefore the Fe can bind two MeIm’s (one on top and the other on bottom), leaving no room for O2.
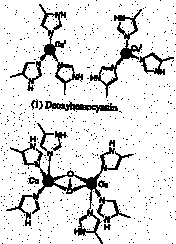
B. Explain the different oxygen binding behavior of the Co(II) prophyrin. From the data given in the problem, we deduce that Co(II) will only bind one axial imidazole, either 1,2-dimethylimidizole or 1-methylimidizole. Thus there is an open coordination site for O2 in either case. C. Why is the P1/2(O2) value unchanged when an additional amount of 1,2-dimethylimidazole is added to the Fe(II) dendritic porphyrin? Since a second 1,2-diMeIm does not bind, it cannot competitively inhibit O2 binding. D. For the two Co(II) dendritic porphyrins, which would have the highest affinity for binding O2 (expressed as K(O2) the equilibrium constant): the complex with the 1,2-dimethylimidazole or with 1-methylimidazole? Whith P1/2(O2) value would be larger? The 1-MeIm ligated system has the highest affinity for binding O2 therefore the P1/2(O2) of 1,2-dimethylimidizole is larger. E. Are any of these complexes diamagnetic; consider either metal, either axial ligand, and O2 as well. Indicate which specific complexes would be diamagnetic. The O2 complex of the Fe(II) porphyrin and the (1-MeIm)2 complex of the Fe(II) porphyrin would be diamagnetic. 2.* (1997 F 9) Hemocyanins, non-heme bimetallic copper proteins, are oxygen carriers in anthropods such as spiny lobster. Below are core structures of deoxy- and oxyhemocyanin. The protein which provides the histidine ligands is not shown. Deoxyhemocyanin has both copper ions in the +1 oxidation state. Oxy-hemocyanin is diamagnetic.
A. Indicate the oxidation state of the copper ions and of the O2 ligand in oxyhemocyanin. Two Cu(II)’s and O22-. B. What single method: mass spec, UV-vis, NMR, microwave, or IR/Raman spectroscopy, would be best suited to verify the oxidation state of the O2 ligand in oxyhemocyanin? Briefly explain your answer–for example, would oxygen isotopes such as 18O2 be useful?
C. Indicate the number of electrons in the copper ions within both deoxy and oxyhemocyanin. Cu(I) in the deoxy is d10; Cu(II) in the oxy is d9. D. Why do you think oxyhemocyanin is diamagnetic The two unpaired electrons, one on each Cu(II), couple by a bonding interaction through the bridging peroxide ligand. 3.* (1997 F 12) Ferrochelatase is an enzyme which inserts iron into protoporphrrin IX; the iron complex is then taken up by hemoglobin. Some people are deficient in this enzyme and have some iron free protoporphyrin IX. Such individuals develop dark lesions on their skin when they are in the sun. Explain the underlying cause of this disease in terms a Chem 31 student would understand. Do you expect this disease to be congenital? Porphyrins are excellent photosensitizers, exciting oxygen from triplet to singlet O2, using light energy (hv). Singlet oxygen is very reactive, and will oxidize funcionalities in the person's cells, etc, leading to the skin lesions. Paramagnetic metals in porphyrins quench singlet oxygen. So, the lack of ferrochelatase, which inserts iron into protoporphyrin IX, is a problem because, without the iron, the singlet oxygen is not quenched. This enzymatic defect is congenital. Enzymes are made by coding in the DNA, so this defect is inherited.
4.* (1995 F 10) Cytochrome-c is a monohemeprotein with two axial ligands bound to iron: a thioether from methionine and an imidazole from histidine. During its function, this iron center cycles between two oxidation states--Fe(II) and Fe(III). The function of this monohemeprotein is not inhibited either by CO nor by cyanide anion. A. Would cytochrome-c be a useful oxygen carrier? Explain your answer. No. The monohemeprotein, because it already has two axial ligands, has no vacant coordination sites to bind O2. B. What is the function of cytochrome-c? It's an electron carrier. C. Cobalt substituted cytochrome-c (Co substituted for Fe) is not known in nature; however, when a synthetic sample of cobalt substituted cytochrome-c was studied, it was found that the rate of electron transfer between the Co(II/III) states is much slower compared with the iron derivative. Explain the rate difference.
Redox changes in iron only involve changes in the nonbonding low spin t2g levels. In cobalt, an electron is removed from an eg antibonding orbitals, so there is a change in bond length. In general, changes in geometry slow down a reaction, so the cobalt reaction is much slower. 5.* (1995 F 12) Cobalt substituted myoglobin (CoMb) binds O2 reversibly as the natural FeMb does. It has been argued that CoMb did not develop in nature because cobalt is much scarcer than iron in the earth’s crust. This is a teleological argument; there may be some other reasons. Some facts about CoMb and FeMb: a) the oxygen adducts CoMbO2 and FeMbO2 have one and no unpaired electrons, repsectively, b) the Co-O bond length in CoMbO2 is longer than the FeO bond length in FeMbO2. The Co(III/II) potential is higher (more positive) than is the Fe(III/II) potential. From these data predict the following and give a reason for your predictions: A. The relative P1/2O2 values for CoMb and FeMb. P1/2O2(CoMb) > P1/2O2(FeMb) because Keq (Fe) > Keq (Co) and B. The relative metal-oxygen (M-O) stretching frequencies (in the infrared or Raman spectra of oxygenated CoMb vs. Fe Mb. The Fe-O bond is shorter and stronger than the Co-O bond. Therefore, the Fe-O stretching frequency would be higher than the Co-O stretching frequency. C. Now consider chromium substituted Mb, CrMb. The Cr(III/II) reduction potential is much lower (it is negative) than either Fe(III/II) or Co(III/II). How do you think the P1/2O2 of CrMb would compare with those of CoMb and FeMb? P1/2O2(CrMb) < P1/2O2(CoMb) < P1/2O2(FeMb) The negative Cr(III)-Cr(II) E° gives CrMb a greater O2 affinity than Co or Fe. D. Predict the number of unpaired electrons in CrMbO2 and justify your prediction.
There are 3 t2g electrons in Cr(III) and one electron from this level goes to form a Ľ bond with (O2-) 6.* (1994 F 10) A. Cytochrome-c is a heme protein with two axial ligands, a thioether and an imidazole (from methionine and histidine residues respectively). The job of cytochrome-c is to act as a single electron carrier. Explain why cytochrome-c is well disigned to carry out this function. Explain the role of spin-states, electron configurations and rates of reaction in your discussion. Compare any difference you expect between the rates of electron transfer for cytochrome-c and an analogue in which the iron atom is replaced by cobalt. Cyt-c must perform rapid outer sphere electron transfer and cycles between the 2+ and 3+ exidation states. Fe(III) is d5: octahedral, low spin. Fe(II) is d6: octahedral, low spin. "extra" electron goes in and out of t2g orbitals…minimal steric effects and bond length changes: fast reaction. Co2+ / Co3+ redox couple cycles between low-spin d6 and high spin d7 (although no difference if low-spin) and there is therefore a large change in electronic structure (occupancy of eg levels)…electron transfer will be much slower. B. When the hemoglobin tetramer binds O2 cooperatively the single axial ligand within each of the four myoglobin-like subunits, an imidazole from the "proximal histidine") moves towards the porphyrin plane upon oxygen binding. The Fe-N bond length decreases. What factor provides for the force behind this movement? Explain as fully as you can. Would you expect a similar movement upon CO binding? Would CO binding exhibit cooperativity? As 5-coordinate Fe(II) [high-spin, s=2, 4 unpaired e-] binds O2 and CO, the resulting complexes are low-spin. The bond lengths shorten dramatically and this spin change provides the force for the movement of the Fe atom. Since the movement occurs for both O2 and CO binding, both will exhibit cooperativity. C. Catalase is a heme protein which catalyzes the decomposition of hydrogen peroxide (H2O2) into water and oxygen. Cyanide anion inhibits catalase but carbon monoxide does not. When isotopically labeled H2O2 (HXXH + HOOH, where X represents 18O and O represents 16O) is allowed to react with catalase, XX and OO are formed but OX is not. Explain the fact that cyanide ion inhibits catalase but carbon monoxide does not. Discuss a plausible mechanism for catalase based on the isotopic labeling experiment. What analytical method would be most appropriate for analysis of isotopically labeled O2? Catalase goes between Fe(III) and the equivalent of Fe(V) (it’s more like Fe(IV) with porphyrin being 1e- oxidized as well) as H2O2 is oxidized and reduced by two 2e- changes. Thus the thermodynamically allowed disproportionation is catalysed…
--> CN- binds Fe(III) and inhibits binding of H2O2 --> stop rxn. --> CO would bind Fe(II) but Fe(II) never occurs --> no binding --> Mass spectrometry would be most appropriate. 7.* (1993 F 7) Carbon monoxide binds strongly to iron(II) porphyrin complexes but not iron(III) porphyrins; conversely cyanide ion binds strongly to iron (III) but not to iron (II) porphyrins. A. Why does CO bind more tightly to iron(II) porphyrins? Explain in one or two sentences; comment on the vCO infrared frequency in this complex. Compare this frequency to that of free CO. The Fe(II)-CO bond is strong because there are strong Ľ-backbonding interactions which strengthen the Fe-C bond. This is much less important for Fe(III)-CO because the charge is higher on the metal. The IR frequency of free CO is higher than the IR frequency of CO in a porphyrin complex because backbonding addes electron density to the p* antibonding orbital of the CO bond, decreasing its strength.B. In what way would CO binding change the reduction potential of an iron(III) porphyrin? Fe(III) does not bind CO, but Fe(II) binds CO strongly, creating a very stable complex ( a complex more stable than Fe(II) by itself.) Since the Fe(II)-CO complex is more stable than Fe(II), the DG of forming the complex is lower (more negative) than the DG of forming Fe(II). Because DG=-nFE, this also means that the reduction potential for Fe(III) and the complex is higher than the reduction potential for Fe(III/II).C. Consider the following heme systems; beside each write CN-, CO, or neither according to whether the function (catalysis, O2 carrier, electron carrier) is expected to be inhibited by CN-, by CO, or by neither.
catalysis, cycles between II and III; binds CO and CN-
electron transport, no free site; neither oxygen carrier, Fe(II); binds CO enzyme, cycles between III and IV; CN- enzyme, cycles between II, III...; CN-, CO
8.* (1992 3 4) What is an enzyme? An enzyme is a protein, a biological catalyst which: •changes the rate of reaction but not the equilibrium conditions (affects kinetics, but not thermodynamics) •is highly substrate-specific (can tell the difference between enantiomers) Some drugs inhibit enzyme binding to substrates. 9.* (1991 F 8) A. Why doesn’t molecular oxygen bond to metals in a 3+ oxidation state? When the metal binds O2, it donates an electron. Therefore, the metal would have to be oxidized to a +4 oxidation state:
This reaction is not favored overall (E°<0, DG > 0)B. How would you distinguish spectroscopically between the superoxide ion and the peroxide ion? Look at the IR stretching frequency: Superoxide (O2-) has a bond order of 1.5, and peroxide (O22-) has a bond order of 1. Superoxide has a stronger bond and would therefore have a higher IR stretching frequency. 10.* (1990 F 14) Cyanide anion binds strongly to iron (III) porphyrin complexes but not to iron (II) prophyrin complexes. Conversely CO binds strongly to Iron (II) but not to iorn (III) porphyrin complexes. A. In what way would cyanide alter the Fe(III)/Fe(II) reduction potential of an iron porphyrin complex? Cyanide forms a stable complex with Fe(III) but not Fe(II). Thus, Fe(III) will not be reduced as easily in the presence of CN-. The reduction potential would be lower. B. The heme enzyme catalase catalyzes the disproportionation of hydrogen peroxide. Write a balanced equation for the overall reaction.
C. Explain the following: CO does not affect the function of catalase but aqueous sodium cyanide inhibits the function of this enzyme. CO binds strongly to Fe(II), and CN- binds strongly to Fe(III). Therefore, the active form of catalse must have Fe(III), not Fe(II). |